目录
1. 衍射峰的强度与哪些因素有关?
2. 棒状、片状、细针状微晶粉末样品做 XRD 重复性很差。制作粉末衍射样品如何避免择优取向?
3. 磁性材料比如NdFeB、NdFeN的粉末,是否会因为磁性的存在会产生择优取向?
4. XRD的光斑是什么样的?
5. XRD 峰整体向右偏移是什么原因造成的?
6. 把样品偏低放置,使样品偏离测角仪中轴大概有1mm,请问衍射峰会怎么变化?
7. XRD衍射强度和衍射峰的宽度与样品颗粒大小,还是与晶体颗粒大小有关?
8. 衍射强度变弱本质的原因是由于晶体颗粒变小,还是样品颗粒变小?
9. 采用X射线进行晶体衍射分析,利用照相法记录衍射花样,当多晶体晶粒细化时,衍射花样将如何变化?当多晶试样中存在宏观应力时,衍射花样的变化情况是怎样?
10. 进行衍射测试时如何选择靶(光管)的原则是什么?现有Cr的多晶试样,我只知道衍射分析时选Cr靶最好,但不知道为什么。
11. 衍射峰左右不对称的原因?
12. 测角仪如何实现θ/2θ倍角转动的?这种装置能够保证严格的倍角同步吗?
13. 薄膜样品用常规方法测试信号不好该怎么办?(什么是掠入射?)
14. 为什么有的XRD数据中,有(200)(400)面,而没有最基本的(100)面数据?或者有(220)而没有(110)?
1. 衍射峰的强度与哪些因素有关?
衍射峰的强度和很多因素有关,比如样品/晶面本身的衍射能力、择优取向、对X射线的吸收能力,还有仪器功率(PXRD功率为40 kV×40 mA,in-situ XRD功率为42 kV×100 mA)、光源强度(与靶头、防发散狭缝、索拉狭缝、单色器等硬件有关)、检测器的灵敏度、样品受光照体积等等;衍射峰强度以Counts计时,与每步扫描时间成正比,以CPS(Counts per Second)计时,与每步扫描时间无关。
2. 棒状、片状、细针状微晶粉末样品做 XRD 重复性很差。制作粉末衍射样品如何避免择优取向?
采用透射法制样:样品研细后,用毛细管法制样,在高分辨透射与PDF散射系统上用透射法测试。研细的样品可随着毛细管的转动,有更多取向的小晶粒被曝光而使取向随机化,因此可以消除择优取向。
毛细管法制样
在反射法中,择优取向是很难完全避免的,只能尽力减少。
在反射法中,采用以下方法可以减少择优取向:
1.样品应当研细(但要适度,要注意样品的晶体结构不要因研磨过度而受到损坏,手捻无颗粒感即可);
2.不要加过多样品,样品成形松散一些;
3.不要在光滑的玻璃板上大力压紧,压样时可以在玻璃板与样品之间隔一张粗糙的纸张;
4.旋转样品;
5.使用带波纹的样品架或采用背装法制样(详见多晶X射线衍射的样品制备 图2 https://iscps.westlake.edu.cn/info/1149/1497.htm)。

设置旋转样品和带波纹的样品架
采用以上方法进行制样和测试后,样品择优取向得到减少,相对峰强与标准PDF卡片一致,如下图(通常择优取向面都是沿某一晶轴的晶面,如001、100)。
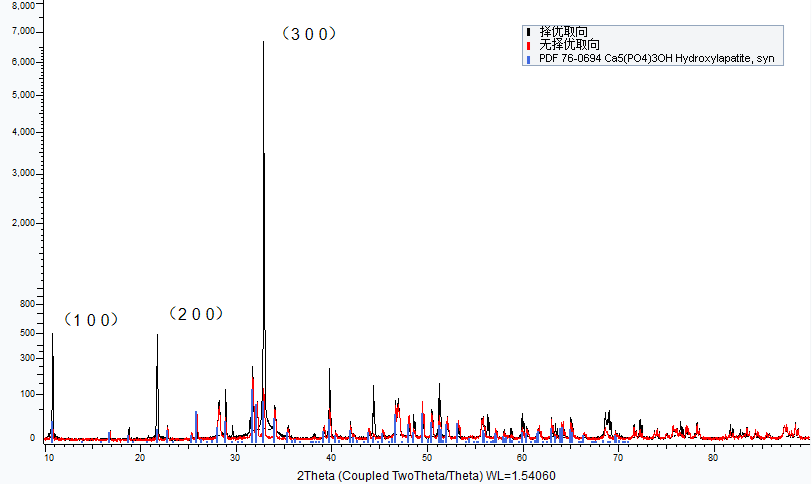
同一粉末样品,制样差异导致的强烈择优取向和无择优取向结果
3. 磁性材料比如NdFeB、NdFeN的粉末,是否会因为磁性的存在会产生择优取向?
磁性材料肯定是最具择优取向的,否则就没有磁性了,制样时应当磨成粉末,旋转样品,可以抑制这种取向趋势。“择优取向”可能会使很多应有的衍射峰不出现。
4. XRD的光斑是什么样的?
PXRD、in-situ XRD、PDF的光源是线焦斑,尺寸为0.4 × 12 mm,照射位置位于样品台中心。
Thin-Film XRD、SCD的光源是点光源,Thin-Film XRD尺寸约直径1 mm,尺寸可以通过准直器调小。
同时,对于所有光源,2θ越低,光斑面积越大,X射线入射样品越浅。
5. XRD 峰整体向右偏移是什么原因造成的?
可能原因有:
1. 峰整体向右偏移说明整体晶面间距减小,可能是离子半径小的元素取代了离子半径大的元素。
2. 制样时样品表面高出样品座平面。

布拉格-布伦塔(Bragg-Brentano)衍射几何以及试样高度位移对峰位的影响
PXRD采用布拉格-布伦塔(Bragg-Brentano)衍射几何,使用平板样品进行测试,因此要求样品的测试面平整致密,并且测试面与样品台的表面高度一致。在正常的测试过程中,样品平面应该与测试圆、衍射圆均相切,样品高度变化后,会引起测试平面与测试圆偏离相切关系,从而导致测得的衍射峰位出现偏移。
如果测试面高于或低于样品台表面,将会引起衍射峰位的偏移与峰形的不对称,峰位偏移与高度偏差存在关系:
Δ2θ = 114.59*s*cosθ/R
其中s =样品位置偏差(mm),R =测角仪半径(mm)(PXRD:R=250 mm)
3. 仪器的零点不准,可以用标样确认。
6. 把样品偏低放置,使样品偏离测角仪中轴大概有1mm,请问衍射峰会怎么变化?
峰整体移向低角度。样品表面偏低测角仪中轴 1 mm,衍射角的测量将产生约 2θ=0.05 °的误差(Cu靶,在2θ=20°附近的位置)。
7. XRD衍射强度和衍射峰的宽度与样品颗粒大小,还是与晶体颗粒大小有关?
晶体颗粒大小。
样品中晶粒越小,衍射峰的峰高和强度越来越低,同时越来越宽,实际上利用X射线衍射峰的宽化对样品的结晶尺寸分析就是根据这个原理(谢乐公式(Scherrer))。
另外,晶粒大小和颗粒大小有关系,但是其各自的含义是有区别的。一颗晶粒也可能就是一颗颗粒,但是更可能的情况是多个晶粒抱到一起,二次聚集,成为颗粒,晶粒之间的接触界面被称为晶界。颗粒不是衍射的基本单位,但是微小的颗粒能产生散射。样品被研磨得越细,散射就越强。对于晶粒,若磨过头,晶体结构就被破坏了,磨成非晶,衍射能力就没有了;若磨得太过,有些峰可能要消失了,而且相邻较近的衍射峰会由于宽化而相互叠加,最终会变成1个或几个“鼓包”,即非晶峰。一般晶面间距大的峰受晶粒细化的影响会明显一些,因为d值大的晶面容易被破坏。
8. 衍射强度变弱本质的原因是由于晶体颗粒变小,还是样品颗粒变小?
晶体颗粒变小。
强度除了和晶粒尺寸有关外,还和晶粒的表面状态有关。一般颗粒越细,其表面积越大,表面层结构的缺陷总是比较严重的。结构缺陷将导致衍射强度降低和衍射峰宽化。XRD 研究的应该是晶粒、晶体的问题,与晶体结构关联的问题,不是样品颗粒的大小问题,谢乐公式算的也是晶粒的大小。样品颗粒的大小要用别的方法测定,例如光散射、X射线散射、电镜等。
9. 采用X射线进行晶体衍射分析,利用照相法记录衍射花样,当多晶体晶粒细化时,衍射花样将如何变化?当多晶试样中存在宏观应力时,衍射花样的变化情况是怎样?
不管一维衍射还是照相法,晶粒细化,会使衍射环变宽。
存在宏观内应力的效应是使得或衍射峰(衍射环)的位置偏移。影响仪器测量结果的分辨率仅仅取决于θ吗?
影响仪器测量结果的分辨率的因素是多方面的:测角仪的半径;X射线源的焦斑尺寸;光学系统的各种狭缝的尺寸;仪器调整情况(2:1关系);采数步宽;样品定位情况等。
10. 进行衍射测试时如何选择靶(光管)的原则是什么?现有Cr的多晶试样,我只知道衍射分析时选Cr靶最好,但不知道为什么。
首先,需要说明产生X射线的两种方式:
1. 对于所有的元素,在高速电子的轰击下都会产生X射线还可能产生其特征X射线。此即光管产生X射线的方式,例如CuKα1的特征波长为1.54059Å。
2. 元素受较高能量的X射线的照射时也能够激发其特征射线,称为二次X射线或荧光X射线,同时表现出对入射X射线强烈的吸收衰减作用。入射靶Kα辐射能量仅略大于样品的特征X射线时,此效应最为明显。
所以,如果样品中的元素的原子序数比靶的元素的原子序数小1至5,就会出现强的荧光散射。由于荧光X射线能量与靶X射线能量接近,所以难以将荧光X射线与衍射X射线区分开来,荧光X射线的强度将叠加在衍射图的背景上,造成很高的背景,不利于衍射图的分析特别是结构精修和定量分析。
因此,选波长(或者说选靶)主要考虑的就是样品中的主要组成元素不会受激发而产生强烈的荧光X射线。例如使用Fe靶分析主要成分元素为Fe、Co、Ni的样品是合适的,而不适合分析含有Mn、Cr、V、Ti的物质;Cu靶不适合于分析有Cr、Mn、Fe、Co、Ni 这些元素的物质。所以,对于Cr是主要组成元素的样品,只能选择Cr靶X射线管。

Fe的含荧光的测试结果和LynxEye-XE去荧光后的测试结果
对于本平台PXRD来说,由于配置了LynxEye-XE-T能量色散一维阵列探测器,能量分辨优于 380 eV,不需要配置Ni Kβ滤片,且不需要额外设置就可以滤除绝大部分的CuKβ和荧光X射线。但是,去除荧光X射线并不意味着解决所有问题。这无法解决Cr、Mn、Fe、Co、Ni对CuKαX射线的吸收,这将使对应物相的衍射强度降低,组分定量发生偏差。因此,含有大量上述元素的样品的定量,依然应当选择其他靶。
11. 衍射峰左右不对称的原因?
衍射仪获得的衍射峰形(精确地说是衍射线的剖面,diffraction line profile)是不对称的,尤其是在低角度区(2θ < 30°)表现更为明显。峰型不对称是由多方面的因素造成的,主要是衍射仪光路的几何因素、仪器的调整状况以及样品的吸收性质(样品对X射线吸收越弱,峰形不对称越明显,所以有机材料在低角度区通常具有更加明显的峰形不对称)等。
12. 测角仪如何实现θ/2θ倍角转动的?这种装置能够保证严格的倍角同步吗?
θ是平板样品与入射X射线的夹角,而 2θ是探测器与入射X射线的夹角,这样设计的目的是为了保证样品在转动中的衍射焦点始终在探测器转动的大圆上。PXRD、in-situ XRD采用立式测角仪,即样品不动,用2个步进电机分别控制θ和 2θ轴的转动。在Coupled TwoTheta/Theta模式中,控制电路控制两个轴按相同转速转动,保证两个轴同步转动,所以入射X射线与平板样品夹角、探测器与平板样品夹角都是θ。
13. 薄膜样品用常规方法测试信号不好该怎么办?(什么是掠入射?)
有部分同学的研究可能会设计到涂层薄膜的研究,比如在玻璃基底上生长电池材料薄膜,这种样品由于绝大多数都是多晶状态,而且厚度有限(通常从几十纳米到微米),如果用常规XRD方法测试,由于在衍射方向薄膜样品厚度非常小,导致参与衍射的晶面非常少,造成样品信号很难测出。这时我们就需要采用掠入射X射线衍射(Grazing Incidence Diffraction, GIXRD或GID)的办法对样品进行测试,掠入射XRD实质是通过X射线小角度照射样品,使更多的晶粒参与衍射,以弥补常规衍射方向厚度的不足,它使X射线束的穿透力被极大的限制,大大提高了分析样品的表面分析灵敏度,且能分析体积相对较大的样品。
14. 为什么有的 XRD数据中,有(200)(400)面,而没有最基本的(100)面数据?或者有(220)而没有(110)?
1. (100)面的角度比较低,可能不在扫描范围内,或淹没在低角度的背景中了。
2. 并非所有晶面的衍射峰都能出现,这取决于“消光规律”。
例如,对于体心立方结构的α-Fe来说,它的消光规律是(h k l)之和为偶数时,衍射峰能够出现,否则会消光。所以α-Fe的衍射数据中有(2 0 0)晶面,而没有(1 0 0)晶面。对于面心立方结构的γ-Fe来说,(h k l)全部为偶数或奇数时,衍射峰能够出现,(h k l)为奇偶混杂时会消光。所以γ-Fe的衍射数据中有(2 0 0)、(2 2 0)晶面,而没有(1 0 0)(1 1 0)晶面。
晶体的“消光规律”决定于结构的对称性,不同的空间群其“消光规律”不同。
来源:网络、Bruker


